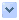地中海貧血(Thalassemia),又稱海洋性貧血,是一種先天的血液疾病,和父母的遺傳有關。 患者的紅血球較脆弱且容易死亡,其帶氧能力亦不足,超過某種程度無法正常生活;在結婚以前健康檢查可以篩選出來,是一種隱性基因遺傳,患者紅血球的體積較正常細胞小,且有時因血紅素含量低較蒼白或呈靶型(target cells)。
病因
地中海貧血症的異常血紅蛋白是在1904年由一個芝加哥醫生發現的,直到1949年人們才通過電泳的方法發現異常血紅蛋白比正常血紅蛋白每個分子多了兩到四個正電荷。胺基酸序列分析證明,地中海貧血症患者的血紅蛋白的一個β亞基第6位胺基酸由正常的谷氨酸突變為異常的頡氨酸,導致血紅蛋白的載氧能力下降。但是另一方面,患有地中海貧血症的患者對瘧疾的抵抗能力要較正常人強,患上瘧疾的地中海貧血症患者,平均存活率顯著高於正常人群,因此在一些瘧疾比較嚴重的地區,如赤道以南非洲,地中海貧血症患者的平均預期壽命反而高於正常人群。
類型
根據血紅蛋白中不同位置的損害可分成兩類:α地中海貧血與β地中海貧血。α地中海貧血是血紅蛋白中的α血紅蛋白鏈有缺損,β地中海貧血則是血紅蛋白中的β血紅蛋白鏈有缺損。鐮刀型紅血球疾病,又名鐮刀型貧血(Sickle Cell Anemia )和地中海貧血不同的是它只發生β血紅蛋白的缺陷。
α地中海貧血
α地中海貧血的患者會在HBA1、HBA2這兩個基因發生異常。在成人造成β蛋白鏈過度製造,在新生兒則是γ蛋白鏈過多。過多的β蛋白鏈形成四聚物(tetramers)使紅血球的攜氧能力降低。
β地中海貧血
β地中海貧血的患者則是在第11號染色體上的HBB基因發生突變。不正常的細胞會製造過量的α蛋白鏈,然後結合在紅血球的細胞膜上造成細胞膜損壞;若其濃度過高,則有形成有毒聚集體(aggregates)的可能。
癥狀
地中海貧血症有隱性、輕度和重度之分。重度患者通常不能存活;而兩個隱性或輕度患者結婚,他們的下一代則有1/4機會患有重度地中海貧血症。相反地,兩者中只有一位是存有地中海貧血症基因的話,不論程度如何,則下一代沒有此問題或只帶有隱性。
由於地中海型貧血的患者缺少正常的血紅素,紅血球攜氧功能差,體內主要造血器官骨髓與次要造血器官肝臟、脾臟均會進行旺盛的造血作用,但造出的紅血球也多半品質不佳,容易被破壞,成為惡性循環。骨髓增生會侵犯周圍的皮質骨,使骨骼較脆弱。旺盛的造血作用會消耗極多的養分與能量,使身體其他部位的養分供需失調。不斷的輸血可以改善貧血的症狀,也可避免過度的造血作用,但血紅素中的鐵質會過度存在身體中,並堆積至各重要器官造成器官病變。
而生活中,地中海貧血患者因血紅素帶氧量不足而影響患者在體力上的差異,患者不適宜進行太激烈的運動。另外亦因為血紅素不足的關係,患者比較容易有頭暈、頭痛甚至腰痛的症狀。
地中海貧血的早期症狀及診斷
如有以下症狀:1.疲勞(感覺疲倦)、衰弱﹔2.膚色蒼白或呈黃色(有如患黃疸病)﹔3.脾臟和肝臟腫大、腹部突出;4.尿液顏色深就要考慮是不是患有地中海貧血,要盡早找醫生做進一步診斷及治療。
診斷地中海貧血,需要進行體格檢查和驗血。體格檢查可以發現肝脾有無腫大。驗血可以了解血紅蛋白水平,如果血紅蛋白水平高於75,那麼就要進行血紅蛋白電泳檢查,看是否有異常血紅蛋白,如果發現了異常血紅蛋白,再進一步進行基因檢查,明確診斷。當你發現你的孩子從小就面色蒼白、腹部有隆起現象、生長發育落後於同齡兒童、鼻子扁平、額骨突出,而你們夫妻雙方又都來自地中海貧血高發地區,就應帶孩子去醫院進行地中海貧血的檢查。
地中海貧血的治療取決於病人類型。如果是輕型地中海貧血或僅僅是地中海貧血基因攜帶者,不會有什麼症狀,也不需要治療。中間型地中海貧血患者根據其貧血嚴重程度不同,需要不同頻率的紅血球輸血。重型地中海貧血患者每2~3週必須接受一次輸血。
文章定位:
人氣(1,505) | 回應(0)| 推薦 (
1)| 收藏 (
0)|
轉寄
全站分類:
不分類 | 個人分類:
醫學常識 |