 STEP 1.
STEP 1. Isolate mouse DRG and place into 15 ml DMEM (serum-free) solution. Centrifuge using 800rpm, at room temperature, for 2 minutes and then remove supernatant.
STEP 2. Add 1 ml DMEM (serum-free) containing 0.125% Type Ia Collagenase (Stock=2.5%, take 50 ul into 950 ul DMEM), and incubate in 37 0C for 1.5 hours.
STEP 3. Add 5 ml DMEM, and then gentled mix for a while. Centrifuge at 800rpm, RT for 2 minutes and then remove supernatant.
STEP 4. Incubate DRG in 1 ml 0.25% trypsin (EDTA-free) at 370C for 15 minutes. Add 5 ml DMEM (with serum) and mix to inactivate enzymatic activity of trypsin. Spin down (800rpm) and remove supernatant. Wash with 10 ml DMEM (serum-free), spin down, and then remove supernatant.
 STEP 5.
STEP 5. Add 2 ml DMEM (serum-free) and triturate DRG with flame polished Pasteur pipette for several( at least 7) times. Rinse the glass pipette with DMEM before contacting the DRG (This procedure will prevent adhesion of DRG on the glass wall).
STEP 6. Transfer to a new tube containing 15 ml DMEM and leave undisrupted for 3 minutes. Fiber debris will fall first in the bottom of the tube. Collect the top 11 ml medium into a new tube without disturbing the debris below. Spin down the DRG at 1000 rpm for 5 minutes.
STEP 7. Resuspend pellet in 2 ml DMEM (containing 1% P/S, 10% FCS, and 2 mM glutamine) and then seed in 3.5 cm dish (coated with 0.1 mg/ml Poly L-lysine).
(1) Mouse embryonic stem cell....medium preparation
(2) Pick-up stable targeting ES cell clone
21th April 2007
Day1.- After 5~7 days of culture with medium containing G418 and ganciclovir , ES cell showed surviving clones. The following protocol describes critical steps in picking up targeting ES clones and preparing genomic DNA from each clone.
Day 6.- Drawing line with marker pen in the bottom of each 10-cm culture dish.
- Mark the position of each surviving clone to save the time in searching clones in the next day.
- Calculate the total number of clones in order to prepare the appropriate amount of MEFs growing in 12-well culture dish. Make MEFs ready to use!
Day 7. - Change Fresh ES medium 1 hour before picking up clones.
- Change ES medium into 5 ml of PBS since serum in ES medium will interfere the proper function of trypsin.
- Prepare 10 ul of trypsin per well in 96-well dish. For beginner without experience, 12 clones/well a time is recommended.
- Place the culture dish with lid open under microscope. Use pipette p20 with filter tip to suck the single ES cell clone. Place the ES cell into one well containing 10 ul of trypsin.
- Place the 96-well into 370c incubator for 2 minutes. At the same time, change MEF medium into ES medium in the 12-well MEFs.
- Add 20 ul PBS into each well and use multi 12 channel pipette loading with 100 ul ES medium to pipette the 12-well trypsin-treated ES clones for at least 30 times in order to resuspand single ES cell .
- Put the 12 channel pipette apart, and recycle each yellow tip (do not mix) with p200 pipette to place single ES clone cell into one 12-well MEFs pre-seeding dish. Label clone number on the lid of each well used.
- Change ES medium without G418 and ganciclovir everyday.
Day 10- Check everyday to see if the confluence of each well reach 50%. Freeze ES clones at this time.
- When ES cells grow up to 50% confluence, suck ES medium out. Wash ES cell with 1 ml/well PBS, and then add 100 ul trypsin and 100 ul PBS into a well. Place the 12 well dish in 370C incubator for 5 minutes.
- Calculate the number of ES cell clones to be frozen and label each freezing tube including date, knockout gene name, experimenter, and ES cell generation.
- Prepare 20% DMSO in ES medium using 50 ml tube (0.5 ml/ tube).
- Pipetting with filter tip for at least 30 times to resuspand single ES cell.
- Add 1 ml ES cell medium into each well to stop trypsin activity. Pre-load 500 ul 20% DMSO in each tube and then take 550 ul of ES cell resuspension into it.
- Cap the tube with fire-burned forceps, and place the tube into cryol-0C freezing container (Nalgene TM) pre-load with room temperature isopropanol in the bottom.
- Place the Cryol-0C freezing container into -800C refrigerator.
- Seeding the left 650 ul ES cell into the same 12 well dish and add extra-500 ul ES medium into each well. Change medium using MEF medium to culture ES cell for genomic DNA extraction.
- Extract ES cells’ genomic DNA when the ES cell reach 80~90% confluence with standard protocol.
接下來因應實驗者對質體DNA在數量和純度上的需求,介紹一種稱為”Banding”的質體DNA的抽取方法。所謂的banding是將質體DNA透過高密度介質氯化銫(CsCl)在超高速離心(ultracentrifuge)下形成分離,質體DNA在超高速離心的氯化銫中形成單一個帶狀故名。因為培養細菌的種類和時程會影響最後質體DNA的產量,故以下以一般實驗室常用的菌種XL1-Blue,吃入質體DNA含Amp選質基因為例,根據實驗時間來依序介紹。
(一)第一天下午五點將單珠菌種移植到3 ml的LB培養液(A+T:使用前加入tetracycline:12.5 ug/ml與ampicilin:50 ug/ml),準備或預借之後所需的實驗材料如下:
滅好菌的LB培養液(500 ml/construct)
測OD值所需比色管
Beckman GSA 10 rotor離心用的250 ml離心管( 兩支 /construct :檢查每支管子的O-ring是否完好)
Chloramphenicol(34 mg/ml:in pure ethanol;2.5 ml/construct)
Solution 1(30 ml /construct)
Solution 2(60 ml /construct)
Solution 3(45 ml /construct)
乾淨的紗布數塊
Isopropanolol(0.6倍體積約81 ml /construct)
1xTE(溶解DNA用,pH=8.0、20 ml /construct)
7.5M Ammonium acetate(4 ml /construct)
Beckman SS-34 rotor與其50 ml離心管(兩支 /construct)
100%酒精(30 ml /construct)
RNase A(10 ul /construct)
CsCl(10 g /construct)
Beckman ultracentrifuge用rotor NVT-65、蓋子(cap)、cap封口把手(有磅數指針)、原廠離心管及封口 焊鐵(見附錄一)
1 ml、5 ml注射針數支、18號、21號針頭、一般15 ml離心管
EtBr(1 ml /construct)
手提式紫外光燈
l-butanol(pure & saturated:含水)
透析用DNA袋子、夾子和裝4公升TE溶液的容器,stir bar等....(二)第二天上午11點將3 ml的菌液取1 ml加到500 ml的 LB培養液(A+T),再放回培養箱進行培養。9~10小時之後進行菌液OD值比色,以乾淨的LB( A+T)為基準做校正,確定細菌生長週期達到plateau(OD值約0.8~1.0之間)。此時加入2.5 ml的 Chloramphenicol(final concentration=170 ug/ml), 再放回培養箱養16小時 。Chloramphenicol的作用在於使細菌停止細胞複製,儘量製造更多的質體DNA,。
(三)第三天下午2點回收細菌,將菌液分裝到250 ml的離心管內,進行4500 rpm(Beckman GSA 10 rotor)、4度C離心20分鐘。倒掉上清液留下下方的pellet,敲打試管或vortex使pellet完全散開,再依序加入S1 30 ml,invert數次,靜置室溫15分鐘;S2 60 ml, gently invert數次,靜置冰上15分鐘;S3 45 ml, gently invert數次, 靜置冰上15分鐘。之後進行5000 rpm(GSA 10 rotor)、4度C離心20分鐘。
(四) 以雙層紗布過濾上清液到另一個乾淨的250 ml離心管,如過濾不乾淨可考慮重覆離心、紗布過濾兩次,確定過濾後的液體澄清,沒有白色沈積物。再加入0.6倍體積的 Isopropanolol(約81 ml), invert數次,靜置冰上15分鐘。之後4度C離心5000 rpm(GSA 10 rotor)20分鐘,倒掉上清液,這時可見瓶底有透明pellet,倒扣瓶子Dry乾液體數分鐘,加入10 ml TE搖晃均勻,最後將10 ml液體(增加回收率可以分4、3、3 ml三次)放到新的50 ml離心管(for SS-34 rotor)。
(五) 加入4 ml 的7.5M Ammonium acetate, 搖晃均勻後靜置冰上10分鐘,4度C離心4500 rpm(SS-34 rotor)10分鐘,此時下方pellet為殘留的細菌染色體與蛋白質,取上清液到新的50 ml離心管(for SS-34 rotor),加入30 ml 100%的酒精,放置負20度C冰箱至少兩小時;再離心 4度C離心10000 rpm(SS-34 rotor)40分鐘;到掉上清液,真空45度C dry乾1小時(可使用保鮮膜封口再行戳洞),之後加蓋保存負20度C冰箱。
(六)解凍後加入10 ml TE溶解下方pellet(增加回收率可以分4、3、3 ml三次),將溶好DNA的TE移到新的15 ml離心管(先取出5 ul當控制組),加入10 ul的RNase A,37度C作用1小時,取5 ul run gel(這步驟可以檢視RNase A是否將RNA清除乾淨)。
(七)精秤CsCl 10克(誤差再0.001g之內)放入50 ml的離心管,將10 ml的TE加入,搖晃至粉末全部溶解;再用5 ml針筒將液體全部加入 Beckman ultracentrifuge 專用離心管( NVT-65 rotor專用),加入時液體由管壁緩慢留下,眼睛檢視絕對不可以有氣泡產生。之後用1 ml針筒將EtBr(有毒物質請小心操作!)加入約0.7~1.0 ml,因為上超高速離心一定要兩邊試管重量相等,所以後果不堪設想!所以加入EtBr的質量提供平衡兩試管重量的好機會(平衡的兩試管誤差應小於0.01克),方可上超高速離心機。
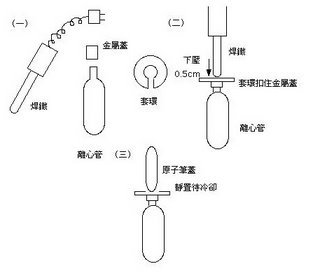
(八)小心用Beckman原廠焊鐵封口(見左圖),封口後可以用手用力壓壓看測試是否完整。封口後invert數下,使EtBr混合均勻,包覆錫泊紙以阻絕光線。上機前在確定兩試管重量相等,方可進行超高速離心。 NVT-65 的 rotor 每支離心管都要加 Cap(with O-ring),用原廠把手鎖緊,確定扭力磅秤讀數約120磅,上機進行超高速離心,離心參數為 20度C、65000 rpm 6小時。
(九)解開 Cap,小心翼翼(勿晃動)將離心管用夾子抽出,鋪好bench紙,打開紫外光燈,此時可見三條主要亮帶(見下圖)先用21號針頭由離心管上方插入(release pressure),在用18號針頭、5 ml針筒由質體DNA的band下方刺入離心管,抽出橘紅色液體約1~2 ml(吸取時可旋轉針頭),放到新的15 ml離心管。

(十)用dropper取兩倍體積(約4 ml)的 Saturated l-butanol(含水),invert混合均勻後室溫離心2000 rpm、3分鐘,用dropper吸掉上清液;再用dropper取兩倍體積(約4 ml)的 Pure l-butanol(不含水), invert混合均勻後室溫離心2000 rpm、3分鐘,用dropper吸掉上清液,這步驟可以去掉大部分的EtBr,準備乾淨的dialysis bag,將質體DNA用p1000移進袋子裡,放到4公升的TE中進行透析,此過程在4度C冰箱中進行,將裝好TE的桶子放在電磁攪拌器上,下方stir bar維持旋轉,6~8小時更換一次乾淨的TE,如此重複3~4次。
這裡要介紹的不是教你如何使用kit,因為鸚鵡學禪的方法在坊間所有販售的kit裡已經有清楚的說明書。如何不用column也可以抽出質體DNA?基因工程實驗常需要挑很多的「克隆(clone)」,挑選的方式不外PCR或colony hybridization這兩種,當實驗者挑到很多可能的「克隆」時,就必須將這些「克隆」進行質體DNA的抽取,以便之後的檢定工作。這裡介紹一個簡便又經濟的抽取方法(特別感謝榮總腎臟科 林堯彬醫師提供)。使用步驟如下:
(1) 將單一菌落(single colony)轉植到1 ml的 LB(事先一實驗設計需要加入特定的抗生素如ampicillin、tetracycline),放在37度C、240 rpm培養16小時。(可以用一般1.5 ml的eppendorf,用小釘子將蓋子口打個小洞)。16小時候將整支試管拿去離心一分鐘(16,000 g)。倒掉上清液,再用震盪器將底部的pellet打散(這步驟很重要,一定要確定打散喔!)。
(2) 加入 100 ul 的溶液S1,invert數下,使之混合均勻。(配方見下附註)。
(3) 加入 200 ul, 的溶液S2,小心輕輕地 invert數下(因為此時細菌的細胞壁被NaOH打破,細菌的染色體DNA外露,為避免斷裂,千萬不可太用力。),靜置室溫5分鐘 。
(4) 加入 200 ul, 的溶液S3,小心輕輕地invert數下後靜置冰上5分鐘。此時細菌的蛋白質和高濃度的醋酸鉀形成白色沈澱。(因為細菌的染色體DNA很長且包附在蛋白質上,會被一起抓下來)。
(5) 4oC離心16,000g 、 5 分鐘,將白色沈澱與上方質體DNA分開。
(6) 準備新的離心管(eppendorf),每隻試管加入1 ml的 酒精(純度至少95%)。
(7) 將步驟(5)的上清液移到酒精中,靜置冰上約30分鐘。
(8) 4oC離心16,000g 、 5 分鐘,倒掉上清液,留下下方pellet。
(9) 用70% 酒精 500 ul清洗pellet。
(10) 4oC離心16,000g 、 5 分鐘,倒掉上清液,用p200儘可能吸乾所有酒精液體。
(11) 倒扣試管室溫下靜置30分鐘,使底部pellet儘量乾燥。之後加入20~50 ul 的TE buffer(pH=8.0)。
理論上,此法所得到的質體DNA,其純度已經可以進行DNA限制酵素(restriction enzyme)的切斷實驗。因為細菌的染色體和蛋白質分離只依賴離心作用,所以質體DNA的純度並不如一般市面上販售的column kit,此方法只建議使用在初步篩選所有可能的「克隆」。如果實驗者挑選到正確的「克隆」,就質體DNA的純度和保存觀點,還是建議將正確「克隆」用column抽取。
附註:
Solution 1(500 ml):秤重Glucose 4.5(g)、加入450 ml純水 stir至完全溶解;在加入0.5M EDTA (pH=8.0)12.5 ml、 1M Tris-HCl (pH=8.0)10 ml,最後補水到500 ml(autoclave後再補滅菌水),使用前加入RNAse(final....100U/ml)4oC保存。
Solution 2(1000 ml):配置0.4N NaOH 500 ml,2% SDS 500 ml(使用無菌純水)。等體積混合兩溶液即可。
Solution 3(200 ml):秤重potassium acetate 58.89(g),加入無菌純水120 ml,再用醋酸glacial acetic acid調整pH值為5.0(約23 ml),最後補無菌純水到200 ml,即為0.5M potassium acetate buffer。 在我們和生技公司購買大腸桿菌勝任細胞時,它通常是以零下-80度C的包裝運送;為了保持這勝任細胞的活性,我們也應將原廠的原始菌種(stock)固定存放在-80度C的冷凍庫中。為了節省經費,這裡介紹一種簡單的製作方法,實驗者可以自行在實驗室裡根據以下步驟進行勝任細胞的製作。製作方法(一)首先,我們準備培養大腸桿菌的培養液LB broth(配方見下表一)、保存勝任細胞的TSB緩衝液(配方見下表二)、預約好光譜儀(spectro-photometer)和低溫離心機( centrifugator)。以LB培養液而言,我們加入秤好的三種原料之後,將整瓶培養液放到滅菌機中進行滅菌(autoclave)。無菌取放3 ml的培養液在養菌專用的試管(Falcon:15 ml),用乾淨的牙籤沾一點原始菌種加入試管,將試管放進37度C、240 rpm震盪的細菌培養箱培養16小時。(二)再準備50 ml LB培養液,倒入滅好菌的錐形瓶。將培養16小時的菌液取100 ul的量(比例約1:500)加入,再放進細菌培養箱培養約3小時。預約光譜儀設定可見光波長為595 nm進行比色,使用乾淨的LB培養液1 ml放入比色管先矯正讀值零點,再將培養3小時的菌液進行比色,確定OD讀值落在0.45~0.55 之間(這讀值代表細菌的生長週期落在其log phase),確定細菌的生長週期之後,之後的操作均在冰上進行。(三)將菌液進行離心(Beckman Rotor),離心的條件為4度C、3000 rpm歷時10分鐘,倒掉上方澄清的培養廢液,再用力敲打離心管使下方沈積的細菌塊鬆散,加入5 ml(置備50支勝任細胞的量)TSB緩衝液(含5% DMSO)。(四)準備冷劑,主要是利用敲碎的乾冰,加入95%的酒精,將試管架的底部浸入約2 cm。在無菌台中操作(laminar flow)、快速地分裝100 ul的菌液到50支乾淨的試管(eppendorf)中,放入冷劑內迅速結凍,之後這些勝任細胞放在-80度C保存。效能檢定與心得討論大腸桿菌勝任細胞的使用效能主要可以分為下列兩點討論:(ㄧ)細胞吃入質體的大小:早期所使用的勝任細胞品係,如 DH5-alpha、XL1-Blue等....大多可以應付10 kb以下大小的質體。但是,隨著基因工程的快速進展,實驗者遇到10 kb 以上的構築實驗機會並不小,所以各家廠商無不專研於研發可以“吃進“更大質體的勝任細胞品係。舉例來說,現在市面上所販售的勝任細胞(如台灣自製的品牌 ECOS 101)大都可以進行10~40 kb的質體操作,這裡必須注意一點,通常質體越大的構築實驗,塞選到正確菌落(colony)的成功機會越小,所以千萬別小看這些市面上販售的「高級」勝任細胞品係,它們真的可以幫你省下很多寶貴的時間。(二)轉型效率(transformation efficiency):轉型效率是影響質體構築實驗成功與否的重要關鍵。用公式來表示轉型效率=所得菌落數目(colony number)/所用的質體DNA質量(ug),換句話說,給予相同質量的質體DNA,一支轉型效率較好的勝任細胞會長出較多的菌落數目,實驗者自製的每一批勝任細胞在使用前都應該進行 轉型效率的測試方可安心使用。這裡介紹一個計算轉型效率的小幫手http://www.sciencegateway.org/tools/transform.htm。一支好的勝任細胞其轉型效率應介於 107 ~ 1010之間(以熱浴方式、質體 pUC-19進行測試)。附帶一提,影響轉型效率的因素還有很多,如質體DNA的品質、質體轉型所採用的方式(heat shock、 chemical or electroporation)、轉型後的培養時間、使用的培養基(LB or SOC medium)、使用質體DNA的大小等.....以台製販售的 ECOS 101而言,採用 42 oC、3.秒熱浴的方式,給予2.7 kb的質體 DNA轉型效率約1.6 ~ 5.5 x 109/ ug,而10.0 kb的質體 DNA轉型效率約4.0 ~ 9.0 x 106/ ug。(引用自官方網站 http://www.yeastern.com)。工欲善其事,必先利其器。製作好用的勝任細胞是成功進行「克隆」實驗的第一步,這裡介紹方法足以應付一般實驗室的需求,實驗者可以自行在實驗室進行製作。如果實驗者發現很多次的嘗試都無法挑到你想要接成的架構質體,可依下列程序思考問題解決:
在我們和生技公司購買大腸桿菌勝任細胞時,它通常是以零下-80度C的包裝運送;為了保持這勝任細胞的活性,我們也應將原廠的原始菌種(stock)固定存放在-80度C的冷凍庫中。為了節省經費,這裡介紹一種簡單的製作方法,實驗者可以自行在實驗室裡根據以下步驟進行勝任細胞的製作。製作方法(一)首先,我們準備培養大腸桿菌的培養液LB broth(配方見下表一)、保存勝任細胞的TSB緩衝液(配方見下表二)、預約好光譜儀(spectro-photometer)和低溫離心機( centrifugator)。以LB培養液而言,我們加入秤好的三種原料之後,將整瓶培養液放到滅菌機中進行滅菌(autoclave)。無菌取放3 ml的培養液在養菌專用的試管(Falcon:15 ml),用乾淨的牙籤沾一點原始菌種加入試管,將試管放進37度C、240 rpm震盪的細菌培養箱培養16小時。(二)再準備50 ml LB培養液,倒入滅好菌的錐形瓶。將培養16小時的菌液取100 ul的量(比例約1:500)加入,再放進細菌培養箱培養約3小時。預約光譜儀設定可見光波長為595 nm進行比色,使用乾淨的LB培養液1 ml放入比色管先矯正讀值零點,再將培養3小時的菌液進行比色,確定OD讀值落在0.45~0.55 之間(這讀值代表細菌的生長週期落在其log phase),確定細菌的生長週期之後,之後的操作均在冰上進行。(三)將菌液進行離心(Beckman Rotor),離心的條件為4度C、3000 rpm歷時10分鐘,倒掉上方澄清的培養廢液,再用力敲打離心管使下方沈積的細菌塊鬆散,加入5 ml(置備50支勝任細胞的量)TSB緩衝液(含5% DMSO)。(四)準備冷劑,主要是利用敲碎的乾冰,加入95%的酒精,將試管架的底部浸入約2 cm。在無菌台中操作(laminar flow)、快速地分裝100 ul的菌液到50支乾淨的試管(eppendorf)中,放入冷劑內迅速結凍,之後這些勝任細胞放在-80度C保存。效能檢定與心得討論大腸桿菌勝任細胞的使用效能主要可以分為下列兩點討論:(ㄧ)細胞吃入質體的大小:早期所使用的勝任細胞品係,如 DH5-alpha、XL1-Blue等....大多可以應付10 kb以下大小的質體。但是,隨著基因工程的快速進展,實驗者遇到10 kb 以上的構築實驗機會並不小,所以各家廠商無不專研於研發可以“吃進“更大質體的勝任細胞品係。舉例來說,現在市面上所販售的勝任細胞(如台灣自製的品牌 ECOS 101)大都可以進行10~40 kb的質體操作,這裡必須注意一點,通常質體越大的構築實驗,塞選到正確菌落(colony)的成功機會越小,所以千萬別小看這些市面上販售的「高級」勝任細胞品係,它們真的可以幫你省下很多寶貴的時間。(二)轉型效率(transformation efficiency):轉型效率是影響質體構築實驗成功與否的重要關鍵。用公式來表示轉型效率=所得菌落數目(colony number)/所用的質體DNA質量(ug),換句話說,給予相同質量的質體DNA,一支轉型效率較好的勝任細胞會長出較多的菌落數目,實驗者自製的每一批勝任細胞在使用前都應該進行 轉型效率的測試方可安心使用。這裡介紹一個計算轉型效率的小幫手http://www.sciencegateway.org/tools/transform.htm。一支好的勝任細胞其轉型效率應介於 107 ~ 1010之間(以熱浴方式、質體 pUC-19進行測試)。附帶一提,影響轉型效率的因素還有很多,如質體DNA的品質、質體轉型所採用的方式(heat shock、 chemical or electroporation)、轉型後的培養時間、使用的培養基(LB or SOC medium)、使用質體DNA的大小等.....以台製販售的 ECOS 101而言,採用 42 oC、3.秒熱浴的方式,給予2.7 kb的質體 DNA轉型效率約1.6 ~ 5.5 x 109/ ug,而10.0 kb的質體 DNA轉型效率約4.0 ~ 9.0 x 106/ ug。(引用自官方網站 http://www.yeastern.com)。工欲善其事,必先利其器。製作好用的勝任細胞是成功進行「克隆」實驗的第一步,這裡介紹方法足以應付一般實驗室的需求,實驗者可以自行在實驗室進行製作。如果實驗者發現很多次的嘗試都無法挑到你想要接成的架構質體,可依下列程序思考問題解決:
(一)切出DNA接點的限制酵素選取是否有錯?是否考慮別的限制酵素在試一次?
(二)DNA的純度夠不夠?一般建議進行接合的材料都要經過 Column或 phenol /chloroform的純化?
(三)是否選用正確的抗生素進行菌落篩選?如雜菌很多建議配置全新的抗生素與培養基使用。
(四)根據你的實驗目的隨時調整你所選用的勝任細胞品係,舉例來說,進行cDNA library的選植實驗就不建議用自製的勝任細胞,如果實驗者欲研究的基因其cDNA表現量就已經不高,他可能會浪費很多的時間還達不到目的。
「克隆」實驗、或是基因工程之所以迷人就是因為「條條道路通羅馬」!這是筆者自身的經驗,幾次嘗試不成之後,表示其中一定有實驗者疏忽或弄錯的地方,這時建議將實驗參數一樣樣進行有「系統」地調整,天無絕人之路,成功常常再不經意的改變之後獲得!
資料來源