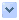From Wikipedia, the free encyclopedia
Enzyme-linked immunosorbent assay (ELISA), also known as an enzyme immunoassay (EIA), is a biochemical technique used mainly in immunology to detect the presence of an antibody or an antigen in a sample. The ELISA has been used as a diagnostic tool in medicine and plant pathology, as well as a quality-control check in various industries. In simple terms, in ELISA, an unknown amount of antigen is affixed to a surface, and then a specific antibody is applied over the surface so that it can bind to the antigen. This antibody is linked to an enzyme, and in the final step a substance is added that the enzyme can convert to some detectable signal, most commonly a colour change in a chemical substrate.
Performing an ELISA involves at least one antibody with specificity for a particular antigen. The sample with an unknown amount of antigen is immobilized on a solid support (usually a polystyrene microtiter plate) either non-specifically (via adsorption to the surface) or specifically (via capture by another antibody specific to the same antigen, in a "sandwich" ELISA). After the antigen is immobilized, the detection antibody is added, forming a complex with the antigen. The detection antibody can be covalently linked to an enzyme, or can itself be detected by a secondary antibody that is linked to an enzyme through bioconjugation. Between each step, the plate is typically washed with a mild detergent solution to remove any proteins or antibodies that are not specifically bound. After the final wash step, the plate is developed by adding an enzymatic substrate to produce a visible signal, which indicates the quantity of antigen in the sample.
Traditional ELISA typically involves chromogenic reporters and substrates that produce some kind of observable color change to indicate the presence of antigen or analyte. Newer ELISA-like techniques utilize fluorogenic, electrochemiluminescent, and real-time PCR reporters to create quantifiable signals. These new reporters can have various advantages including higher sensitivities and multiplexing.[1][2] In technical terms, newer assays of this type are not strictly ELISAs, as they are not "enzyme-linked" but are instead linked to some non-enzymatic reporter. However, given that the general principles in these assays are largely similar, they are often grouped in the same category as ELISAs.
[edit] Applications

ELISA results using S-OIV A
neuraminidase antibody at 1 μg/ml to probe the immunogenic and the corresponding seasonal influenza A
neuraminidase peptides at 50, 10, 2, and 0 ng/ml.
Because the ELISA can be performed to evaluate either the presence of antigen or the presence of antibody in a sample, it is a useful tool for determining serum antibody concentrations (such as with the HIV test[3] or West Nile Virus). It has also found applications in the food industry in detecting potential food allergens such as milk, peanuts, walnuts, almonds, and eggs.[4] ELISA can also be used in toxicology as a rapid presumptive screen for certain classes of drugs.
The ELISA was the first screening test widely used for HIV because of its high sensitivity. In an ELISA, a person's serum is diluted 400-fold and applied to a plate to which HIV antigens are attached. If antibodies to HIV are present in the serum, they may bind to these HIV antigens. The plate is then washed to remove all other components of the serum. A specially prepared "secondary antibody" — an antibody that binds to other antibodies — is then applied to the plate, followed by another wash. This secondary antibody is chemically linked in advance to an enzyme. Thus, the plate will contain enzyme in proportion to the amount of secondary antibody bound to the plate. A substrate for the enzyme is applied, and catalysis by the enzyme leads to a change in color or fluorescence. ELISA results are reported as a number; the most controversial aspect of this test is determining the "cut-off" point between a positive and a negative result.
A cut-off point may be determined by comparing it with a known standard. If an ELISA test is used for drug screening at workplace, a cut-off concentration, 50 ng/mL, for example, is established, and a sample that contains the standard concentration of analyte will be prepared. Unknowns that generate a signal that is stronger than the known sample are "positive." Those that generate weaker signal are "negative."
Doctor Dennis E Bidwell and Alister Voller created the test.
[edit] History
Before the development of the ELISA, the only option for conducting an immunoassay was radioimmunoassay, a technique using radioactively-labeled antigens or antibodies. In radioimmunoassay, the radioactivity provides the signal, which indicates whether a specific antigen or antibody is present in the sample. Radioimmunoassay was first described in a paper by Rosalyn Sussman Yalow and Solomon Berson published in 1960.[5]
Because radioactivity poses a potential health threat, a safer alternative was sought. A suitable alternative to radioimmunoassay would substitute a non-radioactive signal in place of the radioactive signal. When enzymes (such as peroxidase) react with appropriate substrates (such as ABTS or 3,3’,5,5’-Tetramethylbenzidine), a change in color occurs, which is used as a signal. However, the signal has to be associated with the presence of antibody or antigen, which is why the enzyme has to be linked to an appropriate antibody. This linking process was independently developed by Stratis Avrameas and G.B. Pierce.[6] Since it is necessary to remove any unbound antibody or antigen by washing, the antibody or antigen has to be fixed to the surface of the container; i.e., the immunosorbent has to be prepared. A technique to accomplish this was published by Wide and Jerker Porath in 1966.[7]
In 1971, Peter Perlmann and Eva Engvall at Stockholm University in Sweden, and Anton Schuurs and Bauke van Weemen in The Netherlands independently published papers that synthesized this knowledge into methods to perform EIA/ELISA.[8][9]
[edit] "Indirect" ELISA
The steps of "indirect" ELISA follows the mechanism below:-
- A buffered solution of the antigen to be tested for is added to each well of a microtiter plate, where it is given time to adhere to the plastic through charge interactions.
- A solution of non-reacting protein, such as bovine serum albumin or casein, is added to block any plastic surface in the well that remains uncoated by the antigen.
- Next the primary antibody is added, which binds specifically to the test antigen that is coating the well. This primary antibody could also be in the serum of a donor to be tested for reactivity towards the antigen.
- Afterwards, a secondary antibody is added, which will bind the primary antibody. This secondary antibody often has an enzyme attached to it, which has a negligible effect on the binding properties of the antibody.
- A substrate for this enzyme is then added. Often, this substrate changes color upon reaction with the enzyme. The color change shows that secondary antibody has bound to primary antibody, which strongly implies that the donor has had an immune reaction to the test antigen. This can be helpful in a clinical setting, and in R&D.
- The higher the concentration of the primary antibody that was present in the serum, the stronger the color change. Often a spectrometer is used to give quantitative values for color strength.
The enzyme acts as an amplifier; even if only few enzyme-linked antibodies remain bound, the enzyme molecules will produce many signal molecules. Within common-sense limitations, the enzyme can go on producing color indefinitely, but the more primary antibody is present in the donor serum the more secondary antibody + enzyme will bind, and the faster color will develop. A major disadvantage of the indirect ELISA is that the method of antigen immobilization is non-specific; when serum is used as the source of test antigen, all proteins in the sample may stick to the microtiter plate well, so small concentrations of analyte in serum must compete with other serum proteins when binding to the well surface. The sandwich or direct ELISA provides a solution to this problem, by using a "capture" antibody specific for the test antigen to pull it out of the serum's molecular mixture.
ELISA may be run in a qualitative or quantitative format. Qualitative results provide a simple positive or negative result (yes or no) for a sample. The cutoff between positive and negative is determined by the analyst and may be statistical. Two or three times the standard deviation (error inherent in a test) is often used to distinguish positive from negative samples. In quantitative ELISA, the optical density (OD) of the sample is compared to a standard curve, which is typically a serial dilution of a known-concentration solution of the target molecule. For example, if a test sample returns an OD of 1.0, the point on the standard curve that gave OD = 1.0 must be of the same analyte concentration as your sample.
[edit] Sandwich ELISA

A sandwich ELISA. (1) Plate is coated with a capture antibody; (2) sample is added, and any antigen present binds to capture antibody; (3) detecting antibody is added, and binds to antigen; (4) enzyme-linked secondary antibody is added, and binds to detecting antibody; (5) substrate is added, and is converted by enzyme to detectable form.
A less-common variant of this technique, called "sandwich" ELISA, is used to detect sample antigen. The steps are as follows:
- Prepare a surface to which a known quantity of capture antibody is bound.
- Block any nonspecific binding sites on the surface.
- Apply the antigen-containing sample to the plate.
- Wash the plate, so that unbound antigen is removed.
- Apply enzyme linked primary antibodies as detection antibodies that also bind specifically to the antigen.
- Wash the plate, so that the unbound antibody-enzyme conjugates are removed.
- Apply a chemical that is converted by the enzyme into a color or fluorescent or electrochemical signal.
- Measure the absorbency or fluorescence or electrochemical signal (e.g., current) of the plate wells to determine the presence and quantity of antigen.
The image to the right includes the use of a secondary antibody conjugated to an enzyme, though, in the technical sense, this is not necessary if the primary antibody is conjugated to an enzyme. However, use of a secondary-antibody conjugate avoids the expensive process of creating enzyme-linked antibodies for every antigen one might want to detect. By using an enzyme-linked antibody that binds the Fc region of other antibodies, this same enzyme-linked antibody can be used in a variety of situations. Without the first layer of "capture" antibody, any proteins in the sample (including serum proteins) may competitively adsorb to the plate surface, lowering the quantity of antigen immobilized. Use of the purified specific antibody to attach the antigen to the plastic eliminates a need to purify the antigen from complicated mixtures before the measurement, simplifying the assay, and increasing the specificity and the sensitivity of the assay.
A descriptive animation of the application of sandwich ELISA to home pregnancy testing can be found here.
[edit] Competitive ELISA
A third use of ELISA is through competitive binding. The steps for this ELISA are somewhat different than the first two examples:
- Unlabeled antibody is incubated in the presence of its antigen (Sample).
- These bound antibody/antigen complexes are then added to an antigen-coated well.
- The plate is washed, so that unbound antibody is removed. (The more antigen in the sample, the less antibody will be able to bind to the antigen in the well, hence "competition.")
- The secondary antibody, specific to the primary antibody is added. This second antibody is coupled to the enzyme.
- A substrate is added, and remaining enzymes elicit a chromogenic or fluorescent signal.
For competitive ELISA, the higher the sample antigen concentration, the weaker the eventual signal. The major advantage of a competitive ELISA is the ability to use crude or impure samples and still selectively bind any antigen that may be present.
(Note that some competitive ELISA kits include enzyme-linked antigen rather than enzyme-linked antibody. The labeled antigen competes for primary antibody binding sites with your sample antigen (unlabeled). The more antigen in the sample the less labeled antigen is retained in the well and the weaker the signal).
It is common that the antigen is not first positioned in the well.
[edit] Multiple and Portable ELISA (M&P ELISA)(ELISA Reverse in published papers)
A new technique (EP 1 499 894 B1 in EPO Bulletin 25.02.209 N. 2009/09; USPTO 7510687 in USPTO Bulletin 31.03.2009; ZL 03810029.0 in SIPO PRC Bulletin 08.04.2009) uses a solid phase made up of an immunosorbent polystyrene rod with 8-12 protruding ogives. The entire device is immersed in a test tube containing the collected sample and the following steps (washing, incubation in conjugate and incubation in chromogenous) are carried out by dipping the ogives in microwells of standard microplates pre-filled with reagents.
The advantages of this technique are as follows:
- The ogives can each be sensitized to a different reagent, allowing the simultaneous detection of different antibodies and/or different antigens for multi-target assays
- The sample volume can be increased to improve the test sensitivity in clinical (saliva, urine), food (bulk milk, pooled eggs) and environmental (water) samples
- One ogive is left unsensitized to measure the non-specific reactions of the sample
- The use of laboratory supplies for dispensing sample aliquots, washing solution and reagents in microwells is not required, facilitating the development of ready-to-use lab-kits and on-site kits.
[edit] See also
[edit] References
- ^ S. Leng, J. McElhaney, J. Walston, D. Xie, N. Fedarko, G. Kuchel (October 2008). "Elisa and Multiplex Technologies for Cytokine Measurement in Inflammation and Aging Research". J Gerontol a Biol Sci Med Sci 63 (8): 879–884. PMC 2562869. PMID 18772478. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2562869.
- ^ M. Adler, S. Schulz, M. Spengler (2009) Cytokine Quantification in Drug Development: A comparison of sensitive immunoassay platforms. Chimera Biotech. (Report). Retrieved 26 January 2010.
- ^ MedLinePlus. "HIV ELISA/western blot." U.S. National Library of Medicine. Last accessed April 16, 2007. http://www.nlm.nih.gov/medlineplus/ency/article/003538.htm
- ^ U. S. Food and Drug Administration. "Food Allergen Partnership." Last accessed April 16, 2007. http://web.archive.org/web/20080325193553/www.cfsan.fda.gov/~dms/alrgpart.html
- ^ YALOW R, BERSON S (1960). "Immunoassay of endogenous plasma insulin in man". J. Clin. Invest. 39: 1157–75. doi:10.1172/JCI104130. PMC 441860. PMID 13846364. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=441860.
- ^ Lequin R (2005). "Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA).". Clin. Chem. 51 (12): 2415–8. doi:10.1373/clinchem.2005.051532. PMID 16179424.
- ^ Wide L, Porath J. Radioimmunoassay of proteins with the use of Sephadex-coupled antibodies. Biochem Biophys Acta 1966;30:257-260.
- ^ Engvall E, Perlman P (1971). "Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G". Immunochemistry 8 (9): 871–4. doi:10.1016/0019-2791(71)90454-X. PMID 5135623.
- ^ Van Weemen BK, Schuurs AH (1971). "Immunoassay using antigen-enzyme conjugates.". FEBS Letters 15 (3): 232–6. doi:10.1016/0014-5793(71)80319-8. PMID 11945853. .
[edit] External links
==============
好像也會用到這個...
實驗海~!!
貝貝來了~~!!
\^O^/